Key Publications
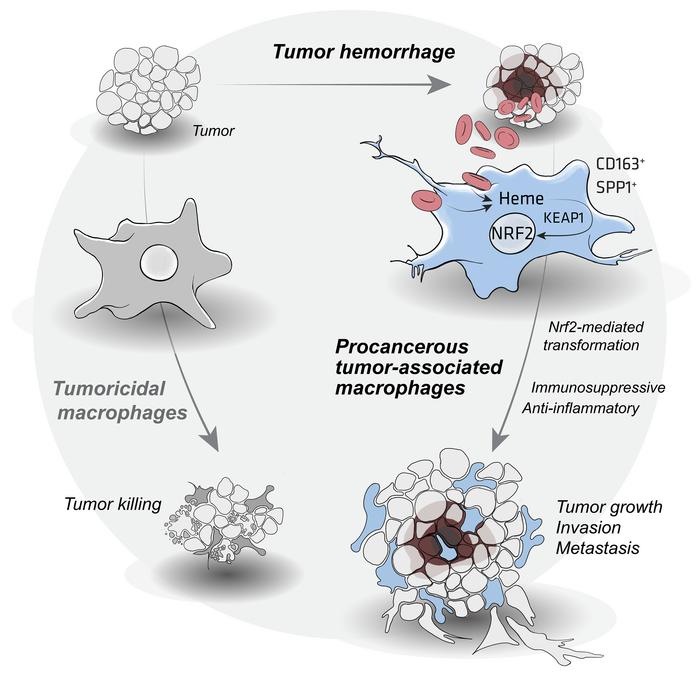
Hemorrhage-activated NRF2 in tumor-associated macrophages drives cancer growth, invasion, and immunotherapy resistance
Schaer DJ, Schulthess-Lutz N, Baselgia L, Hansen K, Buzzi RM, Humar R, Dürst E, Vallelian F. (2024).
Journal of Clinical Investigation.
Abstract
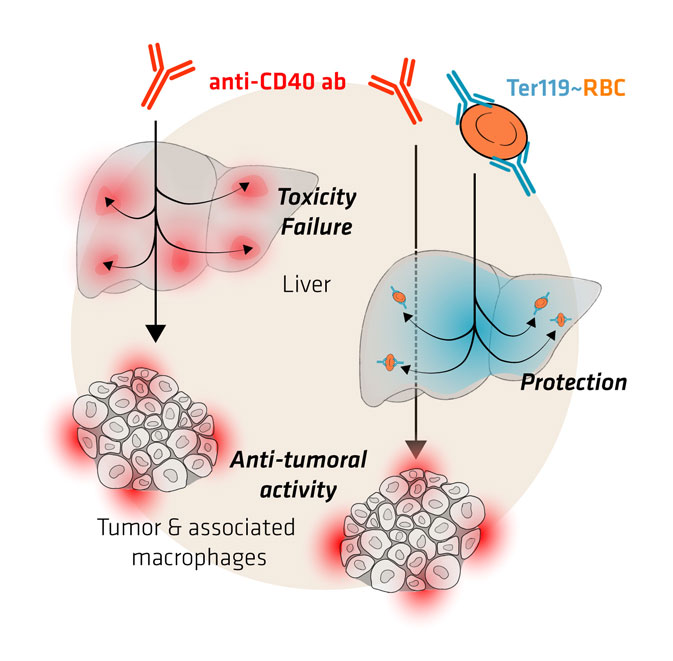
Antibody-induced erythrophagocyte reprogramming of Kupffer cells prevents anti-CD40 cancer immunotherapy-associated liver toxicity
Pfefferlé M*, Dubach IL*, Buzzi RM, Dürst E, Schulthess-Lutz N, Baselgia L, Hansen K, Imhof L, Koernig S, Le Roy D, Roger T, Humar R, Schaer DJ, Vallelian F. (2023).
Journal of ImmunoTherapy of Cancer.
Abstract
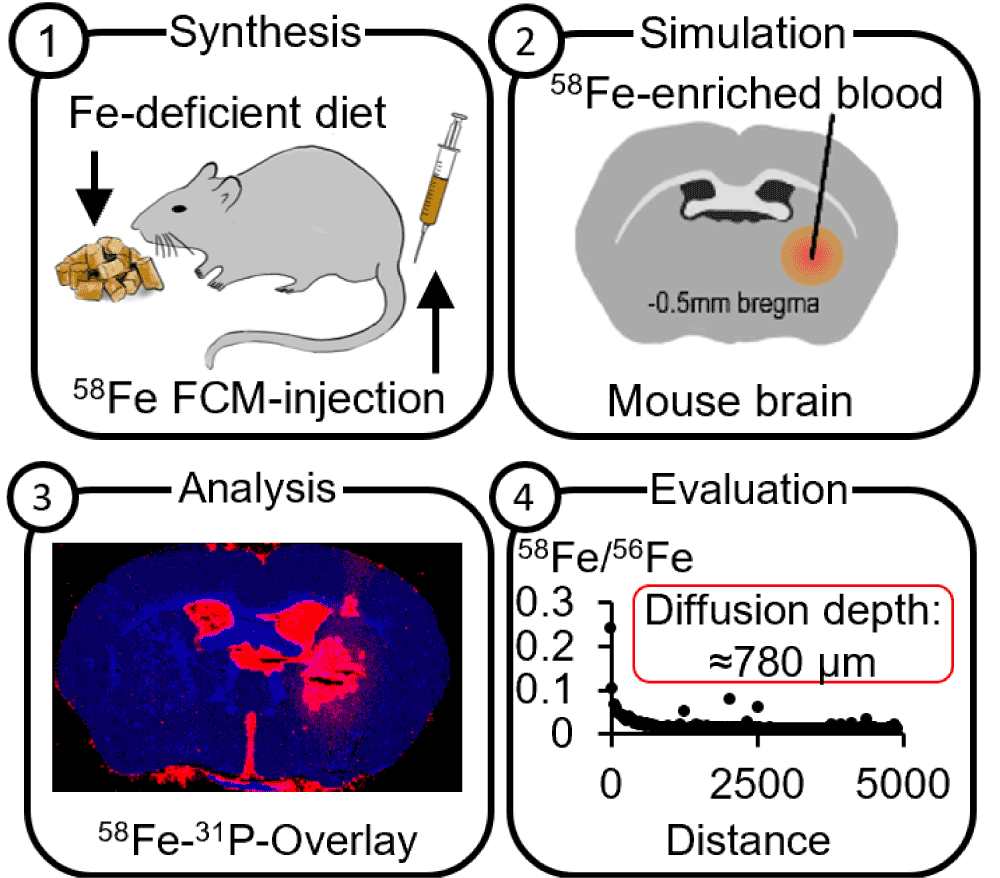
A model to visualize the fate of iron after intracranial hemorrhage using isotopic tracers and elemental bioimaging
Bücker P*, Buzzi RM*, Akeret K, Mosberger L, Richter H, Sperling M, Hugelshofer M, Schaer DJ, Karst U. (2023).
Metallomics.
Abstract

Erythrophagocytes in hemolytic anemia, wound healing, and cancer
Humar R, Schaer DJ, Vallelian F (2022).
Trends in Molecular Medicine.
Abstract
Hemolysis is a ubiquitous pathology defined as premature red blood cell destruction within the circulation or local tissues. One of the most archetypal functions of macrophages is phagocytosis of damaged or extravasated red blood cells, preventing the extracellular release of toxic hemoglobin and heme. Upon erythrophagocytosis, spiking intracellular heme concentrations drive macrophage transformation into erythrophagocytes, leveraging antioxidative and iron recycling capacities to defend against hemolytic stress. This unique phenotype transformation is coordinated by a regulatory network comprising the transcription factors BACH1, SPI-C, NRF2, and ATF1. Erythrophagocytes negatively regulate inflammation and immunity and may modulate disease-specific outcomes in hemolytic anemia, wound healing, atherosclerosis, and cancer. In this opinion article, we outline the known and presumed functions of erythrophagocytes and their implications for therapeutic innovation and research.
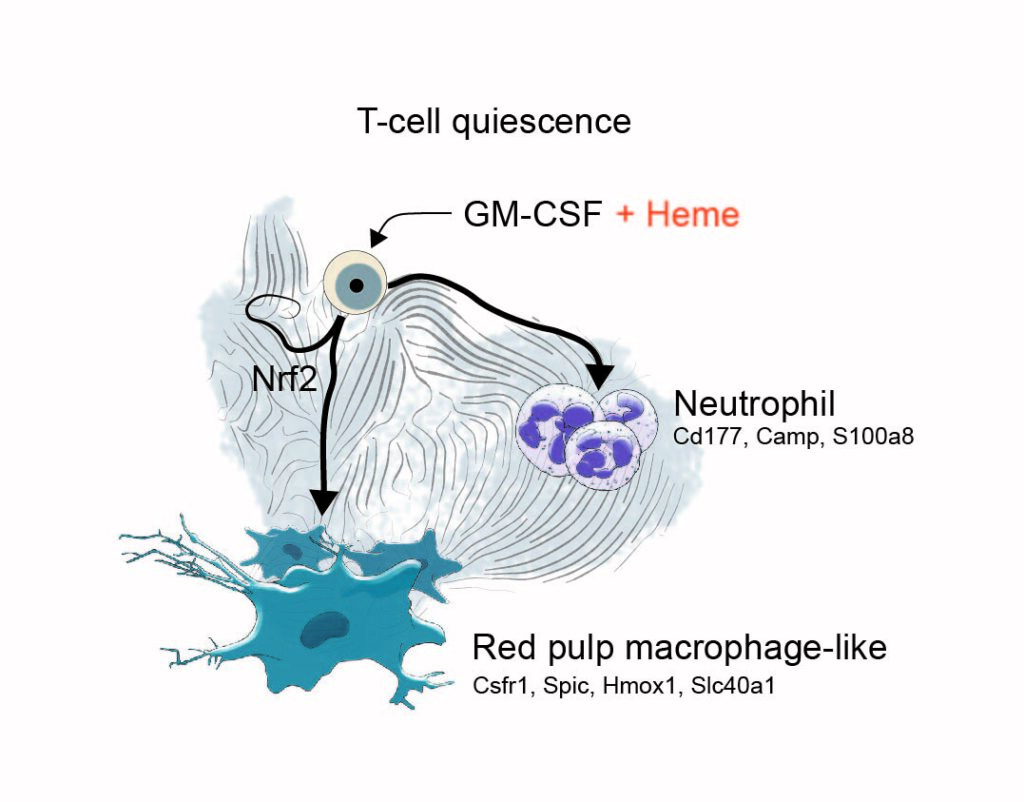
Heme-stress activated NRF2 skews fate trajectories of bone marrow cells from dendritic cells towards red pulp-like macrophages in hemolytic anemia.
Vallelian F, Buzzi RM, Pfefferlé M, Yalamanoglu A, Dubach IL, Wassmer A, Gentinetta T, Hansen K, Humar R, Schulthess N, Schaer CA, Schaer DJ. (2022).
Cell Death & Differentiation.
Abstract
Heme is an erythrocyte-derived toxin that drives disease progression in hemolytic anemias, such as sickle cell disease. During hemolysis, specialized bone marrow-derived macrophages with a high heme-metabolism capacity orchestrate disease adaptation by removing damaged erythrocytes and heme-protein complexes from the blood and supporting iron recycling for erythropoiesis. Since chronic heme-stress is noxious for macrophages, erythrophagocytes in the spleen are continuously replenished from bone marrow-derived progenitors. Here, we hypothesized that adaptation to heme stress progressively shifts differentiation trajectories of bone marrow progenitors to expand the capacity of heme-handling monocyte-derived macrophages at the expense of the homeostatic generation of dendritic cells, which emerge from shared myeloid precursors. This heme-induced redirection of differentiation trajectories may contribute to hemolysis-induced secondary immunodeficiency. We performed single-cell RNA-sequencing with directional RNA velocity analysis of GM-CSF-supplemented mouse bone marrow cultures to assess myeloid differentiation under heme stress. We found that heme-activated NRF2 signaling shifted the differentiation of bone marrow cells towards antioxidant, iron-recycling macrophages, suppressing the generation of dendritic cells in heme-exposed bone marrow cultures. Heme eliminated the capacity of GM-CSF-supplemented bone marrow cultures to activate antigen-specific CD4 T cells. The generation of functionally competent dendritic cells was restored by NRF2 loss. The heme-induced phenotype of macrophage expansion with concurrent dendritic cell depletion was reproduced in hemolytic mice with sickle cell disease and spherocytosis and associated with reduced dendritic cell functions in the spleen. Our data provide a novel mechanistic underpinning of hemolytic stress as a driver of hyposplenism-related secondary immunodeficiency.

Spatial transcriptome analysis defines heme as a hemopexin-targetable inflammatoxin in the brain
Buzzi RM, Akeret K, Schwendinger N, Klohs J, Vallelian F, Hugelshofer M, Schaer DJ. (2022).
Free Radical Biology and Medicine.
Abstract

Modular Platform for the Development of Recombinant Hemoglobin Scavenger Biotherapeutics
Buzzi RM, Owczarek CM, Akeret K, Tester A, Pereira N, Butcher R, Brügger-Verdon V, Hardy MP, Illi M, Wassmer A, Vallelian F, Humar R, Hugelshofer M, Buehler PW, Gentinetta T, Schaer DJ. (2021).
Molecular Pharmaceutics.
https://doi.org/10.1021/acs.molpharmaceut.1c00433
Abstract

Cerebrospinal fluid hemoglobin drives subarachnoid hemorrhage-related secondary brain injury
Akeret K, Buzzi RM, Schaer CA, Thomson BR, Vallelian F, Wang S, Willms J, Sebök M, Held U, Deuel JW, Humar R, Regli L, Keller E, Hugelshofer M, Schaer DJ. (2021).
Journal of Cerebral Blood Flow & Metabolism.
https://doi.org/10.1177/0271678X211020629
Abstract
Secondary brain injury after aneurysmal subarachnoid hemorrhage (SAH-SBI) contributes to poor outcomes in patients after rupture of an intracranial aneurysm. The lack of diagnostic biomarkers and novel drug targets represent an unmet need. The aim of this study was to investigate the clinical and pathophysiological association between cerebrospinal fluid hemoglobin (CSF-Hb) and SAH-SBI. In a cohort of 47 patients, we collected daily CSF-samples within 14 days after aneurysm rupture. There was very strong evidence for a positive association between spectrophotometrically determined CSF-Hb and SAH-SBI. The accuracy of CSF-Hb to monitor for SAH-SBI markedly exceeded that of established methods (AUC: 0.89 [0.85-0.92]). Temporal proteome analysis revealed erythrolysis accompanied by an adaptive macrophage response as the two dominant biological processes in the CSF-space after aneurysm rupture. Ex-vivo experiments on the vasoconstrictive and oxidative potential of Hb revealed critical inflection points overlapping CSF-Hb thresholds in patients with SAH-SBI. Selective depletion and in-solution neutralization by haptoglobin or hemopexin efficiently attenuated the vasoconstrictive and lipid peroxidation activities of CSF-Hb. Collectively, the clinical association between high CSF-Hb levels and SAH-SBI, the underlying pathophysiological rationale, and the favorable effects of haptoglobin and hemopexin in ex-vivo experiments position CSF-Hb as a highly attractive biomarker and potential drug target.

Hemolysis transforms liver macrophages into anti-inflammatory erythrophagocytes
Pfefferlé M, Ingoglia G, Schaer CA, Yalamanoglu A, Buzzi RM, Dubach IL, Tan G, López-Cano EY, Schulthess N, Hansen K, Humar R, Schaer DJ, Vallelian F. (2020).
The Journal of Clinical Investigation.
https://doi.org/10.1172/JCI137282
Abstract
During hemolysis, macrophages in the liver phagocytose damaged erythrocytes to prevent the toxic effects of cell-free hemoglobin and heme. It remains unclear how this homeostatic process modulates phagocyte functions in inflammatory diseases. Using a genetic mouse model of spherocytosis and single-cell RNA sequencing, we found that erythrophagocytosis skewed liver macrophages into a unique anti-inflammatory phenotype that we defined as Marcohigh/Hmoxhigh/MHC-class IIlow erythrophagocytes. This phenotype transformation profoundly mitigated disease expression in a model of an anti-CD40-induced hyperinflammatory syndrome with necrotic hepatitis and in a non-alcoholic steatohepatitis model, representing two macrophage-driven sterile inflammatory diseases. We reproduced the anti-inflammatory erythrophagocyte transformation in vitro by heme-exposure of mouse and human macrophages, yielding a distinctive transcriptional signature that segregated heme-polarized from M1- and M2-polarized cells. Mapping transposase-accessible chromatin in single cells by sequencing (scATAC-seq) defined the transcription factor NFE2L2/NRF2 as a critical driver of erythrophagocytes, and Nfe2l2/Nrf2-deficiency restored heme-suppressed inflammation. Our findings point to a pathway that regulates macrophage functions to link erythrocyte homeostasis with innate immunity.

Haptoglobin Therapeutics and Compartmentalization of Cell-Free Hemoglobin Toxicity
Buehler PW, Humar R, Schaer DJ. (2020).
Trends in Molecular Medicine.
Abstract

Haptoglobin administration into the subarachnoid space prevents hemoglobin-induced cerebral vasospasm
Michael Hugelshofer, Raphael M. Buzzi, Christian A. Schaer, Henning Richter, Kevin Akeret, Vania Anagnostakou, Leila Mahmoudi, Raphael Vaccani, Florence Vallelian, Jeremy W. Deuel, Peter W. Kronen, Zsolt Kulcsar, Luca Regli, Jin Hyen Baek, Ivan S. Pires, Andre F. Palmer, Matthias Dennler, Rok Humar, Paul W. Buehler, Patrick R. Kircher, Emanuela Keller, Dominik J. Schaer. (2019).
The Journal of clinical investigation, 129(12).
https://doi.org/10.1172/JCI130630
Abstract
Delayed ischemic neurological deficit (DIND) is a major driver of adverse outcomes in patients with aneurysmal subarachnoid hemorrhage (aSAH), defining an unmet need for therapeutic development. Cell-free hemoglobin that is released from erythrocytes into the cerebrospinal fluid (CSF) is suggested to cause vasoconstriction and neuronal toxicity, and correlates with the occurrence of DIND. Cell-free hemoglobin in the CSF of patients with aSAH disrupted dilatory NO signaling ex vivo in cerebral arteries, which shifted vascular tone balance from dilation to constriction. We found that selective removal of hemoglobin from patient CSF with a haptoglobin-affinity column or its sequestration in a soluble hemoglobin-haptoglobin complex was sufficient to restore physiological vascular responses. In a sheep model, administration of haptoglobin into the CSF inhibited hemoglobin-induced cerebral vasospasm and preserved vascular NO signaling. We identified 2 pathways of hemoglobin delocalization from CSF into the brain parenchyma and into the NO-sensitive compartment of small cerebral arteries. Both pathways were critical for hemoglobin toxicity and were interrupted by the large hemoglobin-haptoglobin complex that inhibited spatial requirements for hemoglobin reactions with NO in tissues. Collectively, our data show that compartmentalization of hemoglobin by haptoglobin provides a novel framework for innovation aimed at reducing hemoglobin-driven neurological damage after subarachnoid bleeding.

Cell-Free Oxyhemoglobin in Cerebrospinal Fluid After Aneurysmal Subarachnoid Hemorrhage: Biomarker and Potential Therapeutic Target
Hugelshofer M, Sikorski CM, Seule M, Deuel J, Muroi CI, Seboek M, Akeret K, Buzzi R, Regli L, Schaer DJ, Keller E. (2018).
World Neurosurgery, 120, e660-e666.
https://doi.org/10.1016/j.wneu.2018.08.141
Abstract
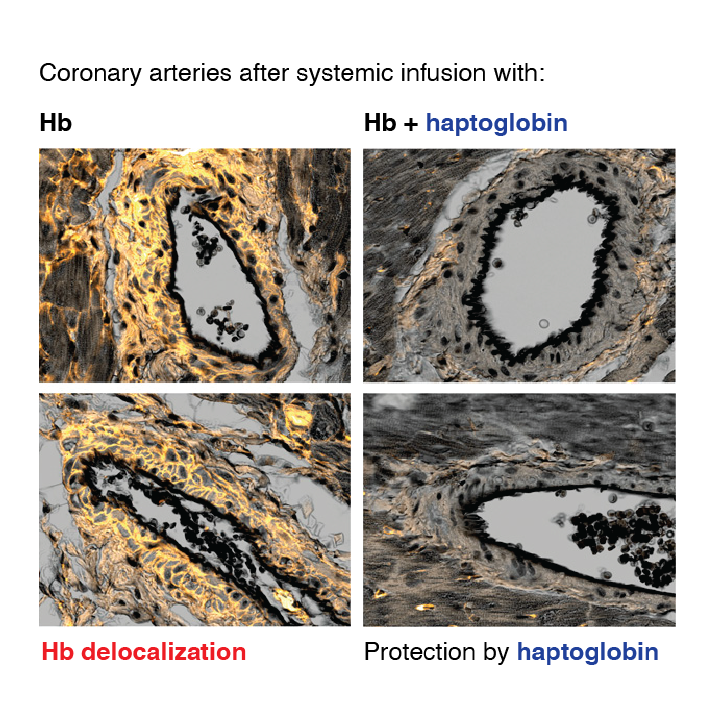
Haptoglobin Preserves Vascular Nitric Oxide Signaling during Hemolysis
Christian A. Schaer, Jeremy W. Deuel, Daniela Schildknecht, Leila Mahmoudi, Ines Garcia-Rubio, Catherine Owczarek, Stefan Schauer, Reinhard Kissner, Uddyalok Banerjee, Andre F. Palmer, Donat R. Spahn, David C. Irwin, Florence Vallelian, Paul W. Buehler, Dominik J. Schaer. (2015).
American journal of respiratory and critical care medicine, 193(10), 1111-1122.
Abstract
Hemolysis occurs not only in conditions such as sickle cell disease and malaria but also during transfusion of stored blood, extracorporeal circulation, and sepsis. Cell-free Hb depletes nitric oxide (NO) in the vasculature, causing vasoconstriction and eventually cardiovascular complications. We hypothesize that Hb-binding proteins may preserve vascular NO signaling during hemolysis. Objectives:
Characterization of an archetypical function by which Hb scavenger proteins could preserve NO signaling during hemolysis.
Methods:
We investigated NO reaction kinetics, effects on arterial NO signaling, and tissue distribution of cell-free Hb and its scavenger protein complexes.
Measurements and Main Results:
Extravascular translocation of cell-free Hb into interstitial spaces, including the vascular smooth muscle cell layer of rat and pig coronary arteries, promotes vascular NO resistance. This critical disease process is blocked by haptoglobin. Haptoglobin does not change NO dioxygenation rates of Hb; rather, the large size of the Hb:haptoglobin complex prevents Hb extravasation, which uncouples NO/Hb interaction and vasoconstriction. Size-selective compartmentalization of Hb functions as a substitute for red blood cells after hemolysis and preserves NO signaling in the vasculature. We found that evolutionarily and structurally unrelated Hb-binding proteins, such as PIT54 found in avian species, functionally converged with haptoglobin to protect NO signaling by sequestering cell-free Hb in large protein complexes.
Conclusions:
Sequential compartmentalization of Hb by erythrocytes and scavenger protein complexes is an archetypical mechanism, which may have supported coevolution of hemolysis and normal vascular function. Therapeutic supplementation of Hb scavengers may restore vascular NO signaling and attenuate disease complications in patients with hemolysis.

Line-selective macrophage activation with an anti-CD40 antibody drives a hemophagocytic syndrome in mice
Ingoglia G, Yalamanoglu A, Pfefferlé M, Dubach IL, Schaer CA, Valkova K, Hansen K, Schulthess N, Humar R, Schaer DJ, Vallelian F. (2020).
Blood advances, 4(12), 2751-2761.
https://doi.org/10.1182/bloodadvances.2020001624
Abstract

Sequestration of extracellular hemoglobin within a haptoglobin complex decreases its hypertensive and oxidative effects in dogs and guinea pigs
Boretti FS, Buehler PW, D’Agnillo F, Kluge K, Glaus T, Butt OI, Jia Y, Goede J, Pereira CP, Maggiorini M, Schoedon G, Alayash AI, Schaer DJ. (2009).
The Journal of clinical investigation, 119(8), 2271-2280.
https://doi.org/10.1172/JCI39115
Abstract
Release of hemoglobin (Hb) into the circulation is a central pathophysiologic event that contributes to morbidity and mortality in chronic hemolytic anemias and severe malaria. These toxicities arise from Hb-mediated vasoactivity, possibly due to NO scavenging and localized tissue oxidative processes. Currently, there is no established treatment that targets circulating extracellular Hb. Here, we assessed the role of haptoglobin (Hp), the primary scavenger of Hb in the circulation, in limiting the toxicity of cell-free Hb infusion. Using a canine model, we found that glucocorticoid stimulation of endogenous Hp synthesis prevented Hb-induced hemodynamic responses. Furthermore, guinea pigs administered exogenous Hp displayed decreased Hb-induced hypertension and oxidative toxicity to extravascular environments, such as the proximal tubules of the kidney. The ability of Hp to both attenuate hypertensive responses during Hb exposure and prevent peroxidative toxicity in extravascular compartments was dependent on Hb-Hp complex formation, which likely acts through sequestration of Hb rather than modulation of its NO- and O2-binding characteristics. Our data therefore suggest that therapies involving supplementation of endogenous Hb scavengers may be able to treat complications of acute and chronic hemolysis, as well as counter the adverse effects associated with Hb-based oxygen therapeutics.

Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification.
Buehler PW, Abraham B, Vallelian F, Linnemayr C, Pereira CP, Cipollo JF, Jia Y, Mikolajczyk M, Boretti FS, Schoedon G, Alayash AI, Schaer DJ. (2009).
Blood (2009) 113 (11): 2578–2586.
Abstract
Detoxification and clearance of extracellular hemoglobin (Hb) have been attributed to its removal by the CD163 scavenger receptor pathway. However, even low-level hydrogen peroxide (H2O2) exposure irreversibly modifies Hb and severely impairs Hb endocytosis by CD163. We show here that when Hb is bound to the high-affinity Hb scavenger protein haptoglobin (Hp), the complex protects Hb from structural modification by preventing α-globin cross-links and oxidations of amino acids in critical regions of the β-globin chain (eg, Trp15, Cys93, and Cys112). As a result of this structural stabilization, H2O2-exposed Hb-Hp binds to CD163 with the same affinity as nonoxidized complex. Endocytosis and lysosomal translocation of oxidized Hb-Hp by CD163-expressing cells were found to be as efficient as with nonoxidized complex. Hp complex formation did not alter Hb’s ability to consume added H2O2 by redox cycling, suggesting that within the complex the oxidative radical burden is shifted to Hp. We provide structural and functional evidence that Hp protects Hb when oxidatively challenged with H2O2 preserving CD163-mediated Hb clearance under oxidative stress conditions. In addition, our data provide in vivo evidence that unbound Hb is oxidatively modified within extravascular compartments consistent with our in vitro findings.

CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin.
Schaer DJ, Schaer CA, Buehler PW, Boykins RA, Schoedon G, Alayash AI, Schaffner A. (2006).
Blood (2006) 107 (1): 373–380.
Abstract
CD163 mediates the internalization of hemoglobin-haptoglobin (Hb-Hp) complexes by macrophages. Because Hp binding capacity is exhausted during severe hemolysis, an Hp-independent Hb-clearance pathway is presumed to exist. We demonstrate that Hb interacts efficiently with CD163 in the absence of Hp. Not only is Hb internalized into an endosomal compartment by CD163 as a result of active receptor-dependent endocytosis; it also inhibits the uptake of Hb-Hp complexes, suggesting a common receptor-binding site. Free Hb further induces heme oxygenase mRNA expression in CD163+ HEK293 cells, but not in CD163– cells. Additional evidence for Hp-independent Hb-CD163 interaction is provided by the demonstration that CD163 mediates the uptake of αα-DBBF crosslinked Hb, a chemically modified Hb that forms minimal Hp complexes. Moreover, certain modifications to Hb, such as polymerization or the attachment of specific functional groups (3 lysyl residues) to the β-Cys93 can reduce or enhance this pathway of uptake. In human macrophages, Hp-complex formation critically enhances Hb uptake at low (1 μg/mL), but not at high (greater than 100 μg/mL), ligand concentrations, lending support for a concentration-dependent biphasic model of macrophage Hb-clearance. These results identify CD163 as a scavenger receptor for native Hb and small-molecular-weight Hb-based blood substitutes after Hp depletion.